Segmentation & event alignment benchmark
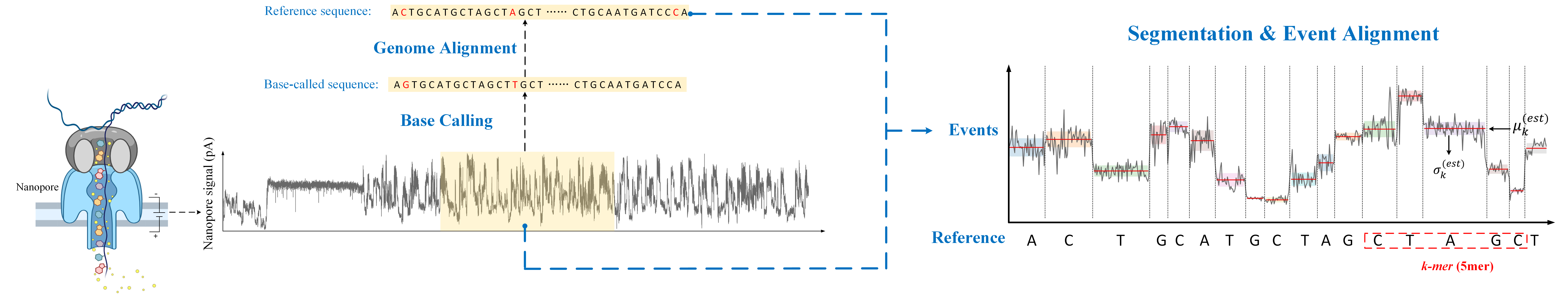
After base calling, we can get a base-called sequence that will align with the reference sequence. The segmentation and event alignment task inputs the raw signal and reference sequence. It would segment the raw signal into small fragments and align it with the reference sequence. So one small fragment and its corresponding nucleotide would be called an "event".
For each fragment of the raw signal, we can get its mean and standard deviation, which are used to evaluate the task.
Segmentation and event alignment benchmark models introduction |
|---|
Tombo re-squiggle
The re-squiggle algorithm is the basis for the Tombo framework. The re-squiggle algorithm takes as input a read file (in FAST5 format) containing raw signal and associated base calls. The base calls are mapped to a genome or transcriptome reference and then the raw signal is assigned to the reference sequence based on an expected current level model. [1]
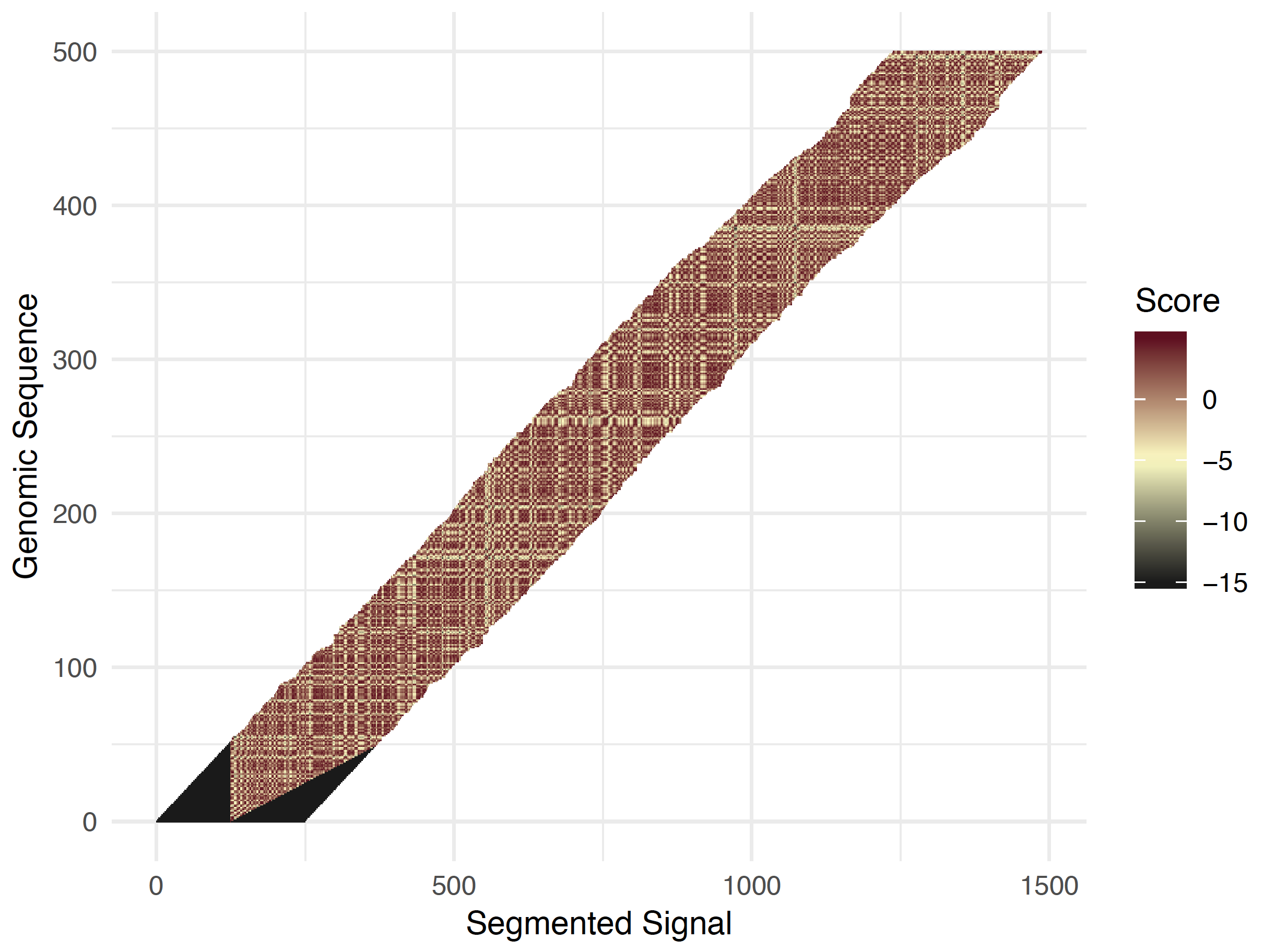
Nanopolish eventalign
The hidden Markov model Nanopolish designed for the consensus problem had 5-mers of a proposed consensus sequence as the backbone of the HMM, with additional states and transitions to handle the skipping/splitting artefacts. Nanopolish used this HMM to calculate a consensus sequence from a set of reads. If we make a reference genome the backbone of the HMM, we can use it to align events to the reference. [2]
SegPore eventalign
SegPore extends the model kmer table by estimating the distribution parameters for both the normal and modified states of each kmer.
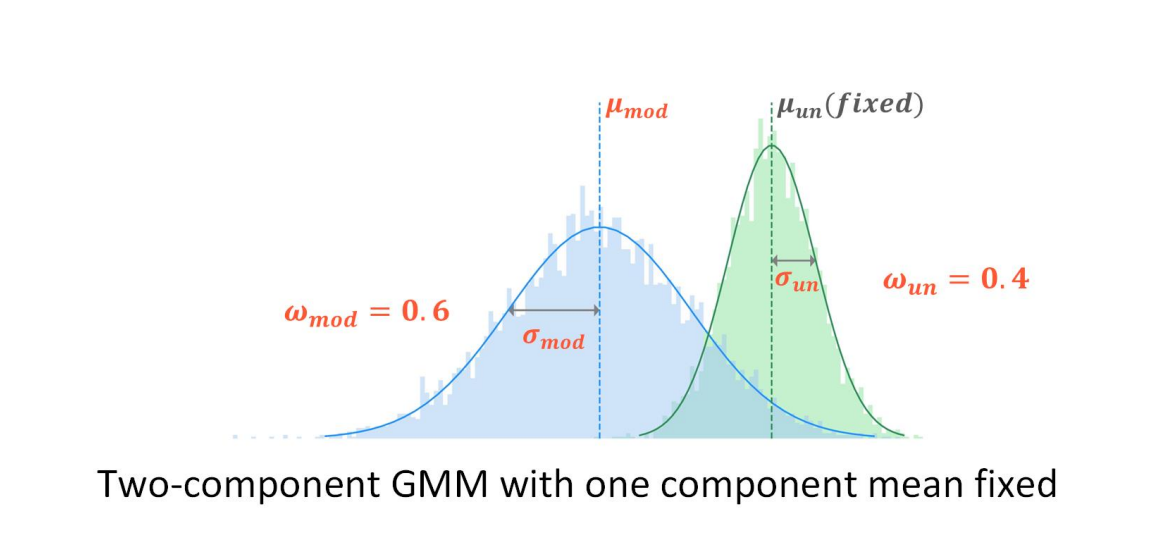
Reference
[1] https://nanoporetech.github.io/tombo/resquiggle.html
[2] https://simpsonlab.github.io/2015/04/08/eventalign
[3] https://www.biorxiv.org/content/10.1101/2024.01.11.575207